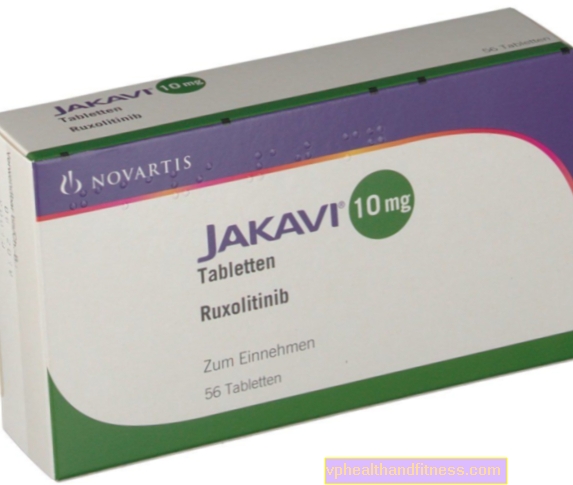
1 tabletta 5 mg, 10 mg, 15 mg vagy 20 mg ruxolitinibet tartalmaz foszfátként. A készítmény laktózt tartalmaz.
| Név | A csomag tartalma | A hatóanyag | Ár 100% | Utoljára módosítva |
| Jakavi | 56 db, asztal | Ruxolitinib | 2019-04-05 |
Akció
Rákellenes gyógyszer, protein-kináz inhibitor. A ruxolitinib a Janus kinázok (JAK), a JAK1 és a JAK2 szelektív inhibitora, amelyek számos citokin és növekedési faktor szignalizációját közvetítik, amelyek fontos szerepet játszanak a vérképzésben és az immunfunkcióban. Nagy áteresztőképesség, jó oldhatóság és gyors kioldódás jellemzi. Orális alkalmazás után gyorsan felszívódik, a Cmax körülbelül 1 órával az adagolás után eléri. A plazmafehérjékhez való kötődés körülbelül 97%, főleg az albuminhoz. A ruxolitinib nem lép át a vér-agy gáton. Főleg a CYP3A4 metabolizálja, további hozzájárulással a CYP2C9. A plazmában a gyógyszer főként változatlan formában és két aktív metabolit formájában van jelen. A ruxolitinib főleg metabolizmus útján eliminálódik. A ruxolitinib átlagos T0,5 eliminációja körülbelül 3 óra, főleg vizelettel és ürülékkel ürül.
Adagolás
Orálisan. A kezelést csak a rákellenes gyógyszerek alkalmazásában jártas orvos kezdeményezheti. A kezelés megkezdése előtt teljes vérképet és fehérvérsejt-vizsgálatot kell végezni. 2–4 hetente teljes vérképet és fehérvérsejt-kenetet kell végezni, amíg az adag stabilizálódik, majd a klinikai indikációktól függően. Kezdeti adag. Myelofibrosis: 15 mg naponta kétszer azoknál a betegeknél, akiknél a vérlemezkeszám 100 000 / mm3 és 200 000 / mm3 között van, és 20 mg naponta kétszer, ha a vérlemezkeszám> 200 000 / mm3. Polycythaemia Vera: 10 mg orálisan naponta kétszer. Korlátozott információ áll rendelkezésre az 50 000 / mm3 és 3 - max. ezeknél a betegeknél az ajánlott kezdő adag naponta kétszer 5 mg, és óvatosan kell emelni. Az adag módosítása. Az adagok a gyógyszer biztonságossága és hatékonysága alapján módosíthatók. A kezelést abba kell hagyni, ha a vérlemezkeszám kevesebb, mint 50 000 / mm3, vagy az abszolút neutrofilszám kevesebb, mint 500 / mm3. A PV-betegek kezelését is fel kell függeszteni, ha a hemoglobinszint 8 g / dl alatt van. Ha a vérképek száma meghaladja ezeket az értékeket, a napi kétszeri 5 mg-os kezelés folytatható, fokozatos emeléssel, a kenetet tartalmazó teljes vérvizsgálat eredményei alapján. A thrombocytopenia miatti kezelés abbahagyásának elkerülése érdekében mérlegelni kell az adag csökkentését, ha a thrombocytaszám 100 000 / mm3 alá csökken. A dózis csökkentését a PV-betegeknél is figyelembe kell venni, ha a hemoglobin értéke 12 g / dl alatt van, és a dózis csökkentése ajánlott, ha a hemoglobin értéke 10 g / dl alatt van. Ha a kezelést nem tartják eléggé hatékonynak, és a vérkép megfelelő, akkor az adag naponta kétszer legfeljebb 5 mg-mal növelhető, legfeljebb max. 25 mg-os adagok naponta kétszer. A kezdő adagot nem szabad a kezelés első négy hetében növelni, és ezt követően nem szabad gyakrabban növelni, mint kéthetes időközönként. Max. a készítmény adagja naponta kétszer 25 mg. Az adag módosítása, ha egyidejűleg erős CYP3A4 inhibitorokat vagy flukonazolt szed. Az egységdózist körülbelül 50% -kal kell csökkenteni, és naponta kétszer kell beadni. Kerülni kell a készítmény egyidejű alkalmazását flukonazollal napi 200 mg-nál nagyobb adagokban. A CYP3A4 erős gátlóival vagy a CYP2C9 és a CYP3A4 enzimek kettős gátlóival történő kezelés során a hematológiai paraméterek, valamint a gyógyszerrel összefüggő mellékhatások jeleinek és tüneteinek gyakoribb (például heti kétszeri) monitorozása ajánlott. A kezelés abbahagyása. A kezelést addig kell folytatni, amíg az előny-kockázat egyensúly pozitív marad, de 6 hónap után abba kell hagyni, ha a lép megkezdése óta nem csökkent a lép mérete vagy tüneti javulás. Javasoljuk, hogy bizonyos fokú klinikai javulást mutató betegek abbahagyják a ruxolitinib alkalmazását, ha a lép megnyúlása az alapvonalhoz képest 40% -os (kb. 25% -os léptágulásnak felel meg), és valódi javulás nem tapasztalható. a betegséggel járó tünetek kapcsán. Speciális betegcsoportok. Enyhe vagy közepesen súlyos vesekárosodásban szenvedő betegeknél nincs szükség különleges dózismódosításra. Súlyos vesekárosodásban szenvedő betegeknél (CCr 3–200 000 / mm3. Egyszeri 20 mg-os adag vagy 2 db 10 mg-os adag 12 órás különbséggel ajánlott azoknak az MF-es betegeknek, akiknél a vérlemezkeszám> 200 000 / mm3. Az ezt követő adagokat (egyszeri adagolás vagy 2 db 10 mg-os adag 12 órás különbséggel) csak hemodialízis napokon szabad beadni, minden dialízis után. Az ESRD-ben és PV-ben szenvedő hemodializált betegek számára javasolt kezdő adag egyetlen 10 mg-os adag vagy minden egyes dialízis után két adag. 5 mg 12 órás időközönként a dialízis után és csak a hemodialízis napján. Ezeket az adagolási javaslatokat szimulálják, és az ESRD-ben szenvedő betegek esetleges dózismódosításait gondosan ellenőrizni kell a biztonságosság és a hatásosság szempontjából. A kezelés alatt álló betegekről nincsenek adatok. peritonealis dialízis vagy folyamatos veno-vénás haemofiltráció. Bármely májkárosodásban szenvedő betegeknél az ajánlott kezdő adag o o a vérlemezkéket kb. 50% -kal kell csökkenteni, és naponta kétszer kell beadni. Az ezt követő adagokat a gyógyszer biztonságossága és hatékonysága alapján kell beállítani. Azoknál a betegeknél, akiknél májkárosodást diagnosztizáltak a készítmény kezelése során, a kezelés megkezdését követő első 6 hétben teljes vérvizsgálatot kell végezni egy kennel legalább 1-2 hét alatt, majd a májfunkció stabilizálása és a vérvizsgálatok után - ha rendelkezésre állnak. klinikai javallatok. Az adag módosítható a citopenia kockázatának csökkentése érdekében. Időseknél nem javasolt további dózismódosítás. A biztonságosság és a hatékonyság 18 év alatti gyermekeknél nem bizonyított. Nincs adat. Adás módja. Vegyük étellel vagy anélkül. Ha kihagy egy adagot, a betegnek nem szabad extra adagot vennie, hanem a következő előírt adagot kell bevennie.
Jelzések
Velőfibrózis. A betegséggel járó splenomegalia vagy a primer csontvelő fibrózisban (más néven krónikus idiopátiás csontvelő fibrózisban is ismert) myelofibrosisban, polycythaemia (hyperaemia) vera által előidézett myelofibrosisban vagy esszenciális thrombocythemia által megelőzött myelofibrosisban szenvedő betegek kezelése. Csomózott csomós kacsa. Polycythemia verában szenvedő felnőtt betegek kezelése, akik rezisztensek vagy intoleránsak a hidroxikarbamid terápiával szemben.
Ellenjavallatok
A készítmény hatóanyagával vagy bármely segédanyagával szembeni túlérzékenység. Terhesség és szoptatás.
Óvintézkedések
A gyógyszer hematológiai mellékhatásokat okozhat, beleértve a thrombocytopeniát, a vérszegénységet és a neutropeniát, ezért a kezelés megkezdése előtt teljes vérvizsgálatot kell végezni fehérvérsejt-kenet alkalmazásával. A kezelést abba kell hagyni, ha a vérlemezkeszám kevesebb, mint 50 000 / mm3, vagy az abszolút neutrofilszám kevesebb, mint 500 / mm3. Megjegyezték, hogy a kezelés megkezdésekor alacsony vérlemezkeszámmal rendelkező betegeknél (3) nagyobb valószínűséggel alakul ki thrombocytopenia a kezelés során. A trombocitopénia általában reverzibilis, és általában a dózis csökkentésével vagy a készítmény ideiglenes elhagyásával kezelhető, de a vérlemezke transzfúzióra lehet szükség a klinikai javallattól függően. Vérszegénységben szenvedő betegeknél vérátömlesztésre lehet szükség, és vérszegénységben szenvedő betegeknél mérlegelni lehet az adag módosítását vagy a kezelés leállítását. Azoknál a betegeknél, akiknek a kezelés kezdetén a hemoglobinszintje 10,0 g / dl alatt van, nagyobb a kockázata a 8,0 g / dl alatti hemoglobinszintnek a kezelés alatt, mint azoknál a betegeknél, akiknél a kiindulási hemoglobinszint magasabb, tehát azoknál a betegeknél, akiknek a kiindulási hemoglobinszintje volt. 10,0 g / dl alatti hemoglobin, a hematológiai paraméterek gyakoribb monitorozása, valamint a készítmény alkalmazásával járó káros hatásokra utaló jelek és tünetek értékelése ajánlott. A neutropenia (abszolút neutrofilszám <500 / mm3) általában reverzibilis volt, és a gyógyszer ideiglenes tartásával kezelhető volt. Teljes vérvizsgálatot kell végezni, amilyen gyakran csak klinikailag javallt, és szükség esetén módosítani kell az adagot. A készítményt kezelt betegeknél súlyos bakteriális, mikobaktériumos, gombás, vírusos és egyéb opportunista fertőzések fordultak elő, ezért meg kell vizsgálni a betegek súlyos fertőzések kockázatát. Az orvosoknak szorosan figyelniük kell a gyógyszert kapó betegeket a fertőzések jeleire és tüneteire, és haladéktalanul megfelelő kezelést kell kezdeményezniük. A kezelést addig nem szabad elkezdeni, amíg már nincs súlyos aktív fertőzés. A myelofibrosis elleni gyógyszert szedő betegeknél tuberkulózist jelentettek, és a kezelés megkezdése előtt a betegeket aktív vagy inaktív (látens) tuberkulózisra kell vizsgálni, a helyi ajánlásoknak megfelelően. A vizsgálatok során figyelembe kell venni a kórtörténetet, a tuberkulózisos betegekkel való esetleges korábbi kapcsolatfelvételt és / vagy a megfelelő szűrővizsgálatokat, például a tüdőröntgen, a tuberkulin teszt és / vagy adott esetben az interferon-y felszabadulás tesztelését. Az orvosoknak tisztában kell lenniük a hamis negatív tuberkulin bőrteszt kockázatával, különösen súlyos betegek vagy immunhiányos betegek esetén. Krónikus HBV-fertőzésben szenvedő betegeknél beszámoltak a hepatitis B vírusterhelésének (HBV-DNS-titer) növekedéséről, az ALT és az AST egyidejű növekedésével vagy anélkül. A gyógyszer hatása a vírusreplikációra krónikus HBV-fertőzésben szenvedő betegeknél nem ismert; a krónikus HBV-betegeket a klinikai irányelvek szerint kell kezelni és ellenőrizni. A zsindely veszélye miatt az orvosoknak utasítaniuk kell a betegeket a korai tünetek és tünetek felismerésére, javasolva a kezelés mielőbbi megkezdését. A Jakavi MF kezelésére történő alkalmazásakor progresszív multifokális leukoencephalopathiáról (PML) számoltak be, ezért az orvosoknak éberen kell figyelniük a PML-re utaló tünetekkel kapcsolatban, amelyeket a betegek nem észlelhetnek (pl. Kognitív, neurológiai vagy szellemi). Figyelemmel kell kísérni a betegeket az új vagy súlyosbodó tünetekre, és ha ilyen tünetek jelentkeznek, mérlegelni kell a beteg neurológushoz történő irányítását vagy megfelelő diagnosztikai ellenintézkedések meghozatalát. PML gyanúja esetén a további kezelést fel kell függeszteni mindaddig, amíg a PML-t kizárják. Ruxolitinib-kezelésben részesült betegeknél beszámoltak a bőr nem melanóma malignus daganatairól (NMSC) (beleértve a bazális sejtes karcinómát, a pikkelyes sejtes karcinómát és a Merkel-sejtes karcinómát), ezeknek a betegeknek a többségét hosszú távú hidroxi-karbamid-terápiával és korábbi NMSC-vel vagy rákot megelőző bőrelváltozásokkal kísérték. Időszakos bőrvizsgálat javasolt azoknál a betegeknél, akiknél fokozott a bőrrák kockázata. A készítmény kezelése a lipidparaméterek növekedésével járt együtt, beleértve az összkoleszterint, a HDL-koleszterint, az LDL-koleszterint és a triglicerideket - ajánlott a lipidszint figyelemmel kísérése és a dyslipidaemia kezelése a klinikai irányelvek szerint. A kezdő adagot csökkenteni kell súlyos vesekárosodásban szenvedő betegeknél. Végstádiumú vesebetegségben és MF-ben hemodialízisben részesülő betegeknél a kezdő dózist a vérlemezkeszám alapján kell meghatározni; a következő adagokat csak a hemodialízis napján szabad beadni, minden egyes hemodialízis befejezése után. Az adagolás további módosítását a gyógyszer biztonságosságának és hatékonyságának gondos figyelemmel kísérésével kell elvégezni. Vesekárosodásban szenvedő betegeknél a kezdő adagot körülbelül 50% -kal kell csökkenteni. A dózis további módosítását a gyógyszer biztonságossága és hatékonysága alapján kell elvégezni. Ha a készítményt erős CYP3A4 inhibitorokkal vagy CYP3A4 és CYP2C9 enzimek kettős inhibitoraival (pl. Flukonazol) együtt kell alkalmazni, az egységdózist körülbelül 50% -kal kell csökkenteni, és naponta kétszer kell beadni. A citoreduktív vagy a vérképző növekedési faktor gyógyszerek egyidejű alkalmazását és a készítményt nem vizsgálták. A kezelés megszakítása vagy abbahagyása után az MF tünetei körülbelül 1 héten belül visszatérhetnek. Vannak olyan esetek, amikor a készítményt abbahagyó kezelés súlyosabb eseményeket tapasztal, különösen más akut társbetegségben szenvedők. Nem ismert, hogy a kezelés hirtelen abbahagyása hozzájárult-e ezekhez az eseményekhez. A készítmény fokozatos csökkentése fontolóra vehető, kivéve, ha a kezelés hirtelen abbahagyására van szükség, bár a dózis csökkentésének hasznosságát nem igazolták. A készítmény laktózt tartalmaz - nem alkalmazható ritka, örökletes galaktóz-intolerancia, Lapp-laktáz-hiány vagy glükóz-galaktóz felszívódási zavarok esetén.
Nemkívánatos tevékenység
A policitémia verában szenvedő betegeknél. Nagyon gyakori: húgyúti fertőzések, CTCAE 3. fokozatú (3) és 3. fokú (50 000 - 25 000 / mm3) vérszegénység, 3. fokozatú neutropenia.(3) és 4 (3) CTCAE, intrakraniális vérzés, gyomor-bélvérzés, egyéb vérzés (beleértve az orrvérzést, a posztoperatív vérzéseket és a haematuriát), puffadás, a CTCAE 3. fokozatú alanin-aminotranszferáz emelkedése ( > 5x - 20x ULN). Nem gyakori: tuberkulózis. Myelofibrosisban szenvedő betegeknél. Nagyon gyakori: bármilyen fokú CTCAE vérszegénység, bármilyen fokú CTCAE vérszegénység, vérzés (bármilyen vérzés, beleértve az intrakraniális és gyomor-bélrendszeri vérzést, véraláfutást és egyéb vérzést, véraláfutást, egyéb vérzést (beleértve az orrvérzést, az eljárás utáni és Haematuria), CTCAE 1. és 2. fokozatú hiperkoleszterinémia, CTCAE 1. fokozatú hipertrigliceridémia, szédülés, CTCAE 1. fokozatú alanin- és aszpartátaminotranszferáz-emelkedések, magas vérnyomás CTCAE 000 - 25 000 / mm3), súlygyarapodás, székrekedés Nem gyakori: CTCAE 3. fokú (3) vérszegénység emelkedett alanin-aminotranszferáz (> 5x - 20x ULN) kezelés után betegeknél MF esetén az MF tünetei, úgymint fáradtság, csontfájdalom, láz, viszketés, éjszakai izzadás, a lép tüneti megnagyobbodása és fogyás jelentkezhetnek. Az MF-vel végzett klinikai vizsgálatokban a teljes MF tüneti pontszám fokozatosan visszaállt a kiindulási értékre a kezelés leállítását követő 7 napon belül.
Terhesség és szoptatás
A készítmény alkalmazása terhesség alatt ellenjavallt. A fogamzóképes nőknek hatékony fogamzásgátló módszereket kell alkalmazniuk a kezelés során, és ha egy nő terhes lesz a kezelés alatt, akkor egyéni kockázat-haszon értékelést kell végezni tanácsadással a magzatra gyakorolt lehetséges kockázatról. Szoptatás alatt nem alkalmazható, ezért a kezelés megkezdésekor a szoptatást abba kell hagyni.
Hozzászólások
Nincs vagy elhanyagolható nyugtató hatása. Azoknak a betegeknek azonban, akiknek szédülést tapasztal a készítmény szedése után, tartózkodniuk kell a gépjárművezetéstől és a gépek kezelésétől.
Interakciók
Interakciós vizsgálatokat csak felnőtteknél végeztek. A ruxolitinib a CYP3A4 és a CYP2C9 által katalizált metabolizmus útján eliminálódik, ezért az ezen enzimek aktivitását gátló készítmények fokozott expozíciót eredményezhetnek a ruxolitinib számára. Ha a gyógyszert erős CYP3A4 inhibitorokkal (például, de nem kizárólagosan, iboceprevir, klaritromicin, indinavir, itrakonazol, ketokonazol, lopinavir / ritonavir, ritonavir, mibefradil, nefazodon, nelfinavir, posakonazol, sakvinavir, telavia egy egysége) alkalmazzák, 50%, és naponta kétszer adják be. A betegeket szorosan figyelemmel kell kísérni (pl. Hetente kétszer) a lehetséges citopenia kialakulása érdekében, és az adagot fokozatosan növelni kell a biztonság és a hatásosság alapján. 50% -os dóziscsökkentést kell fontolóra venni, ha olyan készítményeket alkalmaznak, amelyek a CYP2C9 és a CYP3A4 enzimek kettős inhibitorai (pl. Flukonazol). Kerülni kell a gyógyszer egyidejű alkalmazását a flukonazollal napi 200 mg-nál nagyobb adagokban. A CYP3A4 induktorainak (például, de nem kizárólagosan az avasimibe, a karbamazepin, a fenobarbitál, a fenitoin, a rifabutin, a rifampin (rifampicin), az orbáncfű (Hypericum perforatum) szedésekor a betegeket szorosan figyelemmel kell kísérni, és a biztonságosság és a hatásosság alapján fokozatosan növelni kell az adagot. Nem javasolt az adag módosítása, ha a ruxolitinibet enyhe vagy mérsékelt CYP3A4 inhibitorokkal (például, de nem kizárólagosan, ciprofloxacin, eritromicin, amprenavir, atazanavir, diltiazem, cimetidin) együtt alkalmazzák, a mérsékelt inhibitorokkal történő kezelés megkezdésekor azonban a betegeket szorosan ellenőrizni kell. CYP3A4: A ruxolitinib gátolhatja a bél P-glikoproteint és az emlőrák rezisztencia fehérjét (BCRP), ami megnövelheti ezen transzporterek szubsztrátjainak, például dabigatrán-etexilátnak, ciklosporinnak, rozuvasztatinnak és potenciálisan digoxinnak a szisztémás expozícióját. a gyógyszer monitorozása (TDM) vagy az említett anyagok beadását követő klinikai állapot. Lehetséges, hogy a P-gp és a BCRP potenciális gátlása a bélben minimálisra csökken, ha a gyógyszer beadása közötti időt a lehető leghosszabb ideig tartják. A hematopoietikus növekedési faktorok és a készítmény egyidejű alkalmazását nem vizsgálták. Nem ismert, hogy a Janus-kinázok (JAK) Jakavi általi gátlása csökkenti-e a hematopoietikus növekedési faktorok hatékonyságát, vagy a hematopoietic növekedési faktorok befolyásolják-e a készítmény hatékonyságát. A citoreduktív terápiák és a készítmény egyidejű alkalmazását nem vizsgálták - ezeknek a gyógyszereknek az egyidejű alkalmazásának biztonságossága és hatékonysága nem ismert. A ruxolitinib nem gátolja az orális CYP3A4 szubsztrát midazolám metabolizmusát - ezért nem várható a CYP3A4 szubsztrátok expozíciójának növekedése, ha ezeket a gyógyszereket Jakavi-val együtt adják. A készítmény nem befolyásolja az etinilösztradiolt és a levonorgesztrelt tartalmazó orális fogamzásgátló farmakokinetikáját, ezért az ezt a kombinációt tartalmazó fogamzásgátlók hatékonysága várhatóan nem csökken a ruxolitinib egyidejű alkalmazása során.
A készítmény a következő anyagot tartalmazza: Ruxolitinib
Megtérített gyógyszer: NEM